
[ad_1]
A gene-focused view of micro organism, archaea, eukarya, and viruses.
As described in Strategies, metagenomes from all 4 depths had been co-assembled collectively, genes had been recognized, and normalized protection values had been attained by recruiting the person pattern reads to the assembled contigs. Imply protection values for genes had been extracted, and for exploratory functions these imply protection values for every pattern had been normalized to be out of 1 million (protection per million (CPM)). Genes recognized had been taxonomically categorized and functionally annotated, and right here we break down these outcomes. We additionally embrace data on read-based classification, as a method to doubtlessly determine if any massive biases may need been launched by means of the assembly-to-gene-calling course of, however the outcomes didn’t largely range in any circumstances.
A gene-focused view of Micro organism
Taxonomically categorized throughout the bacterial area, 764,307 distinctive genes had been assembled and recognized, with 461,332, 679,559, 656,264 and 677,165 of them having a protection larger than 0 in Layer 1, Layer 2, Layer 3, and Layer 4, respectively (Tables S2 and S3; Fig. 1A1). Nevertheless, the whole normalized coverages of the bacterial genes decreased with depth (Fig. 1A1). Layer 1 contained the best protection of bacterial genes, with 901,325.3 CPM, whereas Layers 3 and Layer 4 contained the bottom, 864,618.79 and 861,536.72 CPM, respectively. Heatmap and cluster analyses primarily based on Bray Curtis resemblance of the genes are proven in Fig. S2A1. The worldwide similarity was > 40%, with Layer 1 being the least related and clustering out individually; the similarity between Layer 2, Layer 3, and Layer 4 was > 80%. SIMPROF evaluation primarily based on Bray Curtis resemblance at 5% of significance degree detected a big distinction between Layers 3 and Layer 4 (p = 0.001) and Layer 3-Layer 4, and Layer 2 (p = 0.001), and between Layer 3-Layer 4- Layer 2 and Layer 1 (p = 0.001) 32.8% of bacterial genes had been efficiently functionally annotated with KO phrases, comprising 357,984 ± 13,375 CPM throughout the 4 layers (imply ± 1SD; Desk S2). In response to KEGG’s groupings, genes with >= 9 common CPM throughout the 4 layers had been ascribed to 42 metabolic pathways, with essentially the most plentiful being associated to genetic data processing, signaling and mobile processes, carbohydrate metabolism, and vitality metabolism (Desk S2).
Gene-level taxonomic classification throughout the Micro organism detected 44 phyla, 106 households, and 430 species (Fig. 1A2, S3, Desk S3). The dominant phyla had been Cyanobacteria, adopted by Chloroflexi, Proteobacteria, Firmicutes and Bacteroidetes. Cyanobacteria had been dominant in Layer 1, whereas Chloroflexi was dominant in Layer 4. At household degree, Microcoleaceae was essentially the most prevalent household, adopted by Coleofasciculaceae and Oscillatoriaceae. At species degree, the dominant species had been Chloroflexi bacterium, Anaerolineae bacterium, Anaerolineales bacterium, Geitlerinema sp. PCC 9228 and Cyanobacteria bacterium J055. Chloroflexi bacterium, Geitlerinema sp. PCC 9228, Cyanobacteria bacterium J055 and Coleofasciculus chthonoplastes had been the predominant species in Layer 1, whereas Chloroflexi bacterium, Anaerolineae bacterium, Anaerolineales bacterium and Thermoflexia bacterium had been dominant species in Layer 4.

Summaries of normalized gene-level coverages damaged down by area; observe the x-axes range between A1-D1. (A1) Bar plots of the genes recognized in micro organism throughout the uppers 4 layers examined [Layer 1 (0-1 mm from surface), Layer 2(1–2 mm from surface), Layer3 (2–3 mm from surface), and Layer 4 (3-4 mm from surface)]. (A2) Heatmap exhibiting the read-based taxonomic classification of micro organism at species degree with coverages >= 9 throughout the uppers 4 layers examined. (B1) Bar plots of the CPM of the 14,184 genes recognized in archaea throughout the uppers 4 layers examined [Layer 1 (0–1 mm from surface), Layer 2(1–2 mm from surface), Layer 3 (2–3 mm from surface), and Layer 4 (3–4 mm from surface)]. (B2) Heatmap exhibiting the read-based taxonomic classification of archaea at species degree throughout the uppers 4 layers examined. (C1) Bar plots of the CPM of the 547 genes recognized in eucaryote throughout the uppers 4 layers examined [Layer 1 (0–1 mm from surface), Layer 2(1–2 mm from surface), Layer 3 (2–3 mm from surface), and Layer 4 (3–4 mm from surface)]. (C2) Heatmap exhibiting the read-based taxonomic classification of eucaryotes at species degree throughout the uppers 4 layers examined. (D1) Bar plots of the CPM of the 394 genes recognized in viruses throughout the uppers 4 layers examined [Layer 1 (0–1 mm from surface), Layer 2(1–2 mm from surface), Layer 3 (2–3 mm from surface), and Layer 4 (3–4 mm from surface)]. (D2) Heatmap exhibiting the read-based taxonomic classification of viruses at species degree throughout the uppers 4 layers examined.
A gene-focused view of Archaea
For the archaeal area, 14,148 distinctive genes had been recognized with 2,762, 8,338, 13,250, 13,793 having coverages > 0 for Layer 1, Layer 2, Layer 3, and Layer 4, respectively. The very best coverages of genes had been detected in Layer 3 and 4 (Fig. 1B1), which had been > 7 occasions extra plentiful than in Layer 1. The dendrogram in Fig. S2A2 exhibits the heatmap and cluster analyses of samples primarily based on Bray Curtis similarities of the archaeal composition of the group. Layers 3 and 4 had been extra related total, at > 70% similarity, whereas Layer 1 was the least related (Fig. S2A2). In response to SIMPROF evaluation, there was a big distinction within the archaeal gene coverages (primarily based on an alpha worth of 0.05) between Layer 3 and Layer 4 (p = 0.022), Layers 3- Layer 4 and Layer 2 (p = 0.001), and Layers 3- Layer 4- Layer 2 and Layer 1 (p = 0.001). 25.7% of archaeal genes had been efficiently functionally annotated with KO phrases, with these unannotated accounting for 7,792.4 ± 5,215.8 CPM throughout the depths (Desk S4). The 14,148 genes had been categorized into 26 KEGG metabolic pathways (Desk S4). Genetic data processing was essentially the most plentiful pathway, whereas the bottom plentiful pathways had been associated to amino acids, mobile processes, cell motility, and sulfur metabolisms (Desk S4).
Gene-level taxonomic classification of archaea is summarized in Figs. 1B2, S4 and Desk S5. A complete of 20 phyla, 36 households and 496 species had been recognized. The dominant phyla had been Euryarchaeota, adopted by Candidatus Lokiarchaeota, Candidatus Micrarchaeota, Candidatus Woesearchaeota, Candidatus Thorarchaeota. At household degree, Methanosarcinaceae had been the dominant household, adopted by Methanotrichaceae, Methanobacteriaceae and Candidatus Methanoperedenaceae. At species degree, Candidatus Lokiarchaeota archaeon had been the dominant species, adopted by Thermoplasmata archaeon, Candidatus Micrarchaeota archaeon and Candidatus Woesearchaeota archaeon.
A gene-focused view of Eukarya
For the eukaryal area, 547 genes had been detected, with 403, 396, 298, 318 of them having coverages > 0 for Layer 1, Layer 2, Layer 3 and Layer 4, respectively. The best coverages had been recognized in Layer 1, with 1,696.08 CPM (Fig. 1C1). The coverages in Layers 2–4 had been > 6 occasions decrease than in Layer1, with 241.6, 188.6, 257.7 CPM for Layer 2, 3 and 4, respectively. The heatmap and cluster evaluation representing the similarity of the 547 genes with depth is present in Fig. S2A3. The worldwide similarity of the eukaryal group was low, < 5%. Layer 1 represented the least related one, whereas the similarity between Layers 3 and 4 was > 80%. In response to SIMPROF evaluation, there was a big distinction within the protection (primarily based on an alpha worth of 0.05) between Layer 1 and Layer 2-Layer 3-Layer 4 (p = 0.001), and between Layer 2-Layer 3 and Layer 4 (p = 0.001), however not between Layer 3 and Layer 4 (p = 1), the place the similarity was > 80%. 26.7% of eukaryal genes had been efficiently functionally annotated with KO phrases, with these unannotated accounting for 377.6 ± 352.6 CPM throughout the depths (Desk S6). In response to KEGG orthology classification, the 547 genes had been grouped in 11 metabolic pathways (Desk S6). Probably the most plentiful pathways had been genetic data processing and photosynthesis. Overwise, the minor coverages of genes had been associated to the pathways sulfur relay system and quorum sensing (Desk S6).
Gene-level taxonomic classification of eukarya recognized 19 phyla, 119 households and 15 species (Fig. 1C2, S5, Desk S7). Bacillariophyta had been the dominant phyla, adopted by Streptophyta and Ascomycota. At household degree, Thalassiosiraceae had been essentially the most predominant household, adopted by Symbiodiniaceae and Bacillariaceae. At species degree, Thalassiosira pseudonana was the predominant species, adopted by Symbiodinium microadriaticum and Fistulifera solaris.
A gene-focused view of Viruses
835 genes had been assembled and taxonomically categorized as viral, with 451, 474, 791 and 759 of them having a protection larger than 0 in Layers 1, 2, 3, and 4, respectively. The coverages of viral genes elevated with depth, with Layers 3 and 4 having protection values > 3 occasions larger than in Layer 1 (Fig. 1D1). Determine S2A4 exhibits the heatmap and cluster evaluation primarily based on the coverages of the 835 genes recognized. The worldwide similarity between the layers was < 20%, with Layer 1 once more being the least related. A SIMPROF take a look at at 5% of significance degree revealed no significance distinction within the protection of the genes between Layer 3 and Layer 4 (p = 0.35), however detected vital variations between Layers 3-Layer 4 and Layer 2 (p = 0.001), and between Layer 2-Layer 3- Layer 4 and Layer 1 (p = 0.001). 17.9% of recognized viral genes had been efficiently functionally annotated with KO phrases, with these unannotated accounting for 1531.0 ± 1095.9 CPM throughout the depths (Desk S8). KEGG grouped these genes into 10 pathways (Desk S8), the very best coverages had been associated to genetic data processing and phage terminase massive subunit/ phage replication initiation protein.
Determine S6 exhibits the read-based classification of the recognized virome at phyla and household ranges: 3 phyla, 16 households and 149 species had been recognized (Desk S9). The dominant phyla had been Uroviricota, adopted by Nucleocytoviricota and Hofneiviricota. At species degree, essentially the most plentiful species was associated to uncultured Caudoviricetes phage, following by Prokaryotic dsDNA virus sp., uncultured Mediterranean phage uvMED, uncultured Mediterranean phage, Streptomyces phage Brock, Cellulophage phage phi17:1, Podoviridae sp. cty5g4, Marseillevirus LCMAC101 (Fig. 1D2).
Residing in Guerrero Negro microbial mat: micro organism, archaea, and virus genes associated to potential adaptation mechanisms
Right here we give attention to recovered genes associated to antibiotic and multidrug resistance, heavy steel toxicity, oxidative injury genes, chilly, warmth and phage shock proteins, UV-radiation stress genes, salinity and desiccation stress circumstances. We recovered 6477 distinctive genes annotated with these capabilities that had been categorized throughout the bacterial area and 44 throughout the archaeal area (Fig. 2, Tables S10, S11, S12, S13, S14, S15, S16 and S17). Furthermore, one gene (associated to UV-DNA injury endonuclease Desk S18) was recovered that was categorized as originating from a virus. In response to KEGG, these genes had been categorized inside membrane transport (76), signaling and mobile processes (488), antimicrobial resistance genes (245), sign transduction (394), genetic data processing (4220), metabolism (758), transport and catabolism (24), replication and restore (10) and vitality metabolism (301) (Fig. 2). The presence of those genes might have implications for adaptability and resilience to emphasize circumstances, as has been beforehand described in one other microbial mat4.
Genes associated to potential adaptation mechanism in Micro organism
For Micro organism, 6477 genes associated to potential environmental adaptation had been detected (Fig. 2A and supplementary Tables S10–S16). In response to KEGG orthology classification, the 6477 genes had been categorized inside membrane transport (76), signaling and mobile processes (480), antimicrobial resistance genes (245), sign transduction (393), genetic data processing (3834), metabolism (749), transport and catabolism (24), replication and restore (370) and vitality metabolism (301): 564 distinctive genes had been associated to multidrug resistance protein/multidrug efflux pump (Desk S10); 86 distinctive genes had been detected for a number of antibiotic resistance protein; 280 genes had been detected for fluoroquinolone, catechol, tetracycline, fosmidomycin, quaternary compound, tetracycline and vancomycin resistance protein; 92 distinctive genes had been recognized to resistance to arsenical, copper, mercuric, tellurite and zinc (Desk S11); 25 genes had been associated to heavy steel (Desk S12); 410 concerned in coping with oxidative stress (Desk S13); 237 genes had been associated to chilly shock proteins (Desk S14); 283 genes had been recognized as warmth shock proteins (Desk S14); 281 genes for phage shock protein (Desk S14); 997 genes had been associated to UV injury: excinuclease ABC subunit ABC, DNA helicase II /ATP-dependent DNA helicase PcrA, UV DNA injury endonuclease, and ATP-dependent DNA helicase UvsW (Desk S15).; and 3222 genes had been related to desiccation and salinity circumstances (Desk S16): 1329 genes had been annotated as RNA polymerase sigma-70 issue, ECF subfamily (rpoE), 301genes for F-type H + /Na + -transporting ATPase subunit beta (ATPF1B, atpD), 281 genes for chaperonin GroEL (groEL, HSPD1), 251 genes for chaperonin GroES (groES, HSPE1), 259 genes for molecular chaperone GrpE (GRPE), 121 genes for molecular chaperone DnaJ (dnaJ), 116 genes for molecular chaperone DnaK (dnaK, HSPA9), 15 genes for trehalose 6-phosphate synthase (otsA), 35 genes for trehalose 6-phosphate phosphatase (otsB), 81 genes for osmoprotectant transport system substrate-binding protein (opuC), 113 genes for glycine betaine/proline transport system ATP-binding protein (proV), 106 genes for glycine betaine/proline transport system permease protein (proW), 206 genes for glycine betaine/proline transport system substrate-binding protein (proX) and eight genes for L-ectoine synthase (ectC). Abstract of those genes and their normalized coverages are confirmed in Fig. 2A.

(A) Heatmap exhibiting the coverages of doubtless adaptation-relevant genes current in micro organism throughout the uppers 4 layers examined. (B) Heatmap exhibiting the coverages of doubtless adaptation-relevant genes current in archaea throughout the uppers 4 layers examined. (C) Heatmap exhibiting the UV gene current in virus throughout the uppers 4 layers examined.
Total, layer 1 contained the very best coverages of genes concerned in antibiotic and multidrug resistance (pumps ATP-binding cassette, subfamily B, multidrug efflux pump and membrane fusion protein, multidrug efflux system), resistance to metals (arsenic, copper resistance protein, tellurite, tellurium and tetracycline genes), heavy steel, oxidative stress genes (superoxide dismutase, Cu–Zn household, superoxide dismutase Fe, Mn household and superoxide oxidase) and phage and warmth shock proteins. Then again, deeper layers contained the best coverages of genes associated to multidrug resistance genes (MFS transporter, ACDE household, multidrug resistance protein, multidrug resistance protein, MATE household and outer membrane protein, multidrug efflux system), zinc resistance gene, superoxide reductase gene, chilly shock protein and genes related to UV-resistance/restore (uvrA, uvrB, uvrC and uvrD genes). Genes concerned with desiccation and salinity stress circumstances had related coverages throughout layers, detecting the best coverages for genes to encoded the manufacturing of exopolysaccharides (rpoE) and molecular chaperones (GroES, GroEL, DnaJ and DnaK). Deeper dialogue to clarify their relevance within the context of potential ecological variations of microbial mats is detailed in Part “Dialogue”.
Genes associated to potential adaptation mechanism in Archaea
Within the archaeal area, 44 genes recognized within the metagenome information had been associated to emphasize circumstances: 2 for multidrug resistance, 4 for a number of antibiotic resistance, 8 for interactions with oxidative genes, 16 shock proteins and 14 for UV genes (Fig. 2B, Desk S17). Layer 3 and 4 contained the best coverages of multidrug resistance genes, antibiotic, oxidative genes (superoxide reductase and superoxide dismutase), chilly and phage shock proteins and UV genes. Then again, antibiotic resistance genes weren’t recovered in Layer 1 or 2; whereas warmth shock protein hsIJ was larger than htpX and the best coverages had been present in Layer 2. In response to KEGG orthology classification, the 44 genes had been categorized inside signaling and mobile processes (8), sign transduction (1), genetic data processing (18), metabolism (9), replication and restore (8). Deeper dialogue to clarify their relevance within the context of potential ecological variations of microbial mats is detailed in Part “Dialogue”.
Interlinking between the genes current in micro organism, archaea and viruses utilizing phylogenetic evaluation
To additional study the distribution of genes associated to potential adaptative mechanisms when it comes to their evolutionary relatedness, we constructed phylogenetic bushes. A complete of 12 bushes had been constructed for the genes: MFS transporter, ACDE household, multidrug resistance protein (KO ID K08221), small multidrug resistance pump (KO ID K03297), superoxide dismutase, Fe–Mn household (KO ID K04564), superoxide reductase (KO ID K05919), warmth shock protein (KO ID K03799), chilly shock protein (KO ID K03704), phage shock protein (KO ID K03973), excinuclease ABC subunit A (KO ID K03701), excinuclease ABC subunit B (KO ID K03702), excinuclease ABC subunit C (KO ID K03703), DNA helicase II ATP dependent DNA helicase (KO ID K03657) and UV DNA injury endonuclease (KO ID K13281). These genes had been current in micro organism, archaea and viruses domains as comply with: for KO ID K08221: 14 sequences had been current in micro organism and 1 in archaea; for KO ID K03297: 21 sequences had been detected in micro organism and 1 in archaea; for KO ID K04564: 171 sequences had been current in micro organism and 1 in archaea; for KO ID K05919: 107 sequences had been present in micro organism and seven in archaea; for KO ID K03799: 106 sequences had been current in micro organism and 4 in archaea; for KO ID K03704: 221 sequences had been present in micro organism and seven in archaea; for KO ID K03973: 114 sequences had been current in micro organism and a pair of in archaea; for KO ID K03701: 362 sequences had been current in micro organism and 5 in archaea; for KO ID K03702: 193 sequences had been current in micro organism and 1 in archaea; for KO ID K03703: 203 sequences had been present in micro organism and three in archaea; for KO ID K03657: 214 had been recognized in micro organism and 1 in archaea; for KO ID K13281: 17 sequences had been detected in micro organism, 2 in archaea and 1 in viruses. Figures 3, 4, 5 and 6 present the phylogenetic relationships between domains; the whole bushes are within the supplementary materials Figs. S7–S15. And see Desk 1 and the corresponding supplemental tables for gene IDs and full amino-acid sequences.
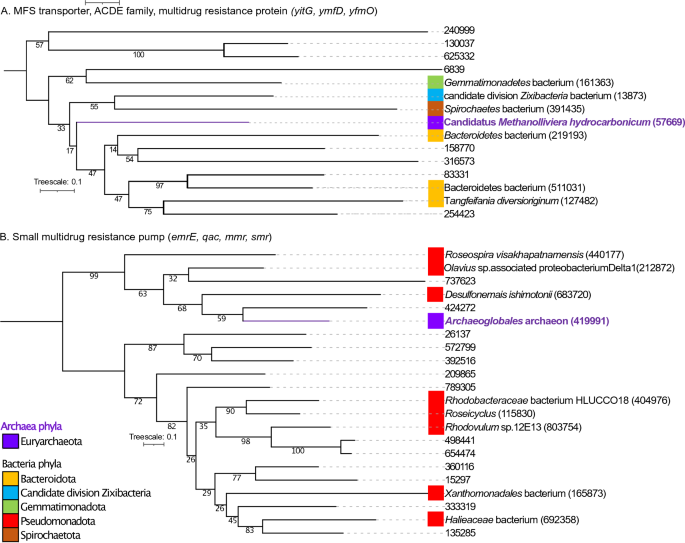
(A) Mid-point rooted phylogenetic tree primarily based on amino-acid sequences detected in our research from MFS transporter, ACDE household, multidrug resistance protein (yitG, ymfD, yfmO). (B) Phylogenetic tree primarily based on amino-acid sequences from small multidrug resistance pump (emrE, qac, mmr, smr). Amino-acid sequences from micro organism (black coloration), amino acid sequences from archaea (purple coloration). For constructed the tree, 37 sequences had been included within the phylogenetic evaluation (15 sequences for yitG, ymfD, yfmO genes and 22 sequences for emrE, qac, mmr, smr genes). The sequences had been aligned with Muscle and the tree was generated with IQTREE2 with 1000 bootstraps, with auto-model choice through the built-in ModelFinder.
Determine 3 exhibits the phylogenetic tree of MFS transporter, ACDE household, multidrug resistance protein (yitG, ymfD, yfmO; 3A) and small multidrug resistance pump (emrE, qac, mmr, smr; 3B). For ACDE household, multidrug resistance protein (yitG, ymfD, yfmO) (Fig. 3A), the gene ID 57669 current in archaea belonging to Candidatus Methanolliviera hydrocarbonicum was between clades holding genes categorized as sourced from the next bacterial lineages: Spirochaetes bacterium (gene ID 391435), Candidate division Zixibacteria bacterium (gene ID 13873), Bacteroidetes bacterium (genes IDs 219193, 511031), Tangfeifania diversioriginum (gene ID 127482) and the genes IDs 158770, 316573, 83331 and 254423. With respect small multidrug resistance pump (emrE, qac, mmr, smr) (Fig. 3B), the gene ID 419991 current in Archaeoglobales archaeon shaped a clade with 4 genes current in micro organism: the genes IDs 424272, 737623, gene ID 683720 current in Desulfonemais himotonii and gene ID 212872 current in Olavius sp. related proteobacterium Delta1.
Determine 4 and Fig. S7–S8 present the phylogenetic bushes for superoxide dismutase, Fe–Mn household (SOD2) (Fig. 4A and S7) and superoxide reductase (dfx) (Fig. 4B and S8). For SOD2, the gene ID 550693 current in archaea shaped a clade with a gene ID 201256 current in micro organism. For dfx gene, the gene ID 612886 current in archaea shaped a clade with 3 genes current in micro organism lineages: two current in Deltaproteobacteria (genes IDs 56860, 26015) and one current in micro organism gene ID 612887. The gene ID 571069 current in Candidatus Micrarchaeota was intently associated to genes current in Deltaproteobacteria (gene ID 59597) and Desulfobacteraceae (gene ID 241743). Lastly, 5 genes current in archaea with genes IDs 323416, 494295, 151464, 732097, 733106 presents in Thermoplasmatales and Thermoplasmata shaped a clade. This clade was intently associated to a clade shaped by the bacterial lineages Candidatus Cloacimonas sp. (411781), Peptoclostridium litorale (685247), Clostridia (847882), Planctomycetes bacterium (315242) and the gene IDs 40055, 525801, 693963, 411780, 6209.
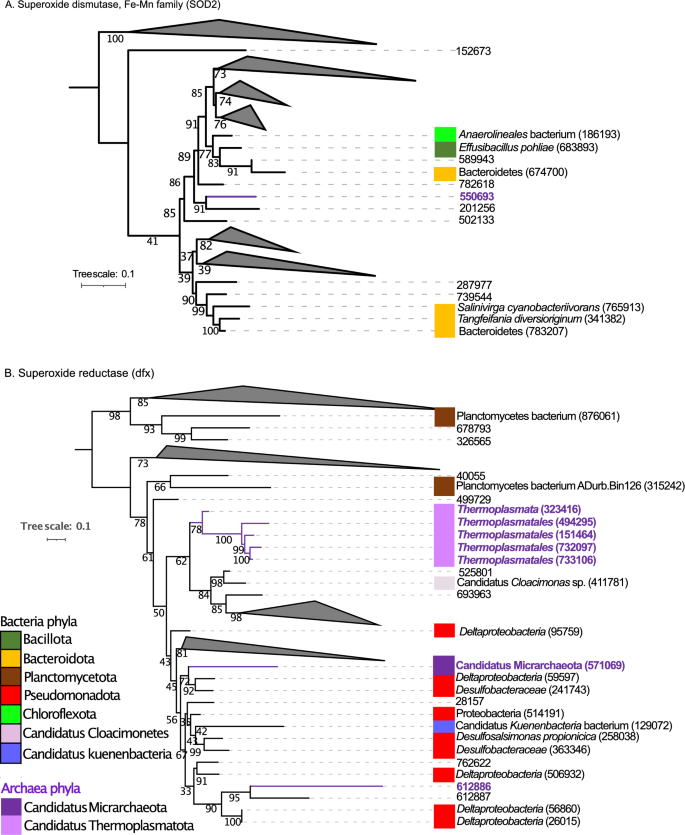
(A) Mid-point rooted phylogenetic tree primarily based on amino-acid sequences detected in our research from superoxide dismutase, Fe–Mn household (SOD2). (B) Phylogenetic tree primarily based on amino-acid sequences from superoxide reductase (dfx). Amino-acid sequences from micro organism (black coloration), amino acid sequences from archaea (purple coloration). For constructed the tree, 286 sequences had been included within the phylogenetic evaluation (172 sequences for SOD2 gene and 114 sequences for dfx gene). The sequences had been aligned with Muscle and the tree was generated with IQTREE2 with 1000 bootstraps, with auto-model choice through the built-in ModelFinder.
Determine 5 and Figures S9–S11 present the phylogenetic bushes similar to warmth, chilly and phage shock protein. For warmth shock protein HtpX (htpx) (Fig. 5A and S9), the gene IDs 443088 and 222562 from the archaea lineages of Thermoplasmata and Candidatus Micrarchaeota shaped a clade with a gene recovered from a Deltaproteobacteria (gene ID 17526). The genes IDs 350980 and 60713 current in archaea deeply branched in between clades holding bacterial genes from Planctomycetes (563474, 523502, 102322), Phycisphaerales (754540), candidate division Zixibacteria (73164) and Desulfofustis sp (446773). Concerning to chilly shock protein (cspA) (Fig. 5B and S10), the genes ID 806860, 245087, 649029, 199423 and 224539 detected from the archaeal lineages of Thermoplasmata, Candidatus Aenigmarchaeota archaeon and Thermoplasmatales shaped a clade with two genes current in Chloroflexi bacterium (genes IDs 641093, 589131, 623971, 155751). The genes ID 454595 and 191917 presents in Candidatus Woesearchaeota and Euryarchaeota had been in a deeply branching clade with genes from Planctomycetota (gene ID 76796) and Bacteroidetes (genes IDs 20000, 628969). For phage shock protein C (pcpc) (Fig. 5C and S11), the gene ID 259327 current in archaea shaped a clade with genes from Bacteroidetes bacterium (gene ID 585054). The gene ID 254336 current in Methanomas siliicoccalesa shaped a clade with bacterial genes from Alphaproteobacteria bacterium (gene ID 125844) and Bacteroidetes bacterium (gene ID 585054).
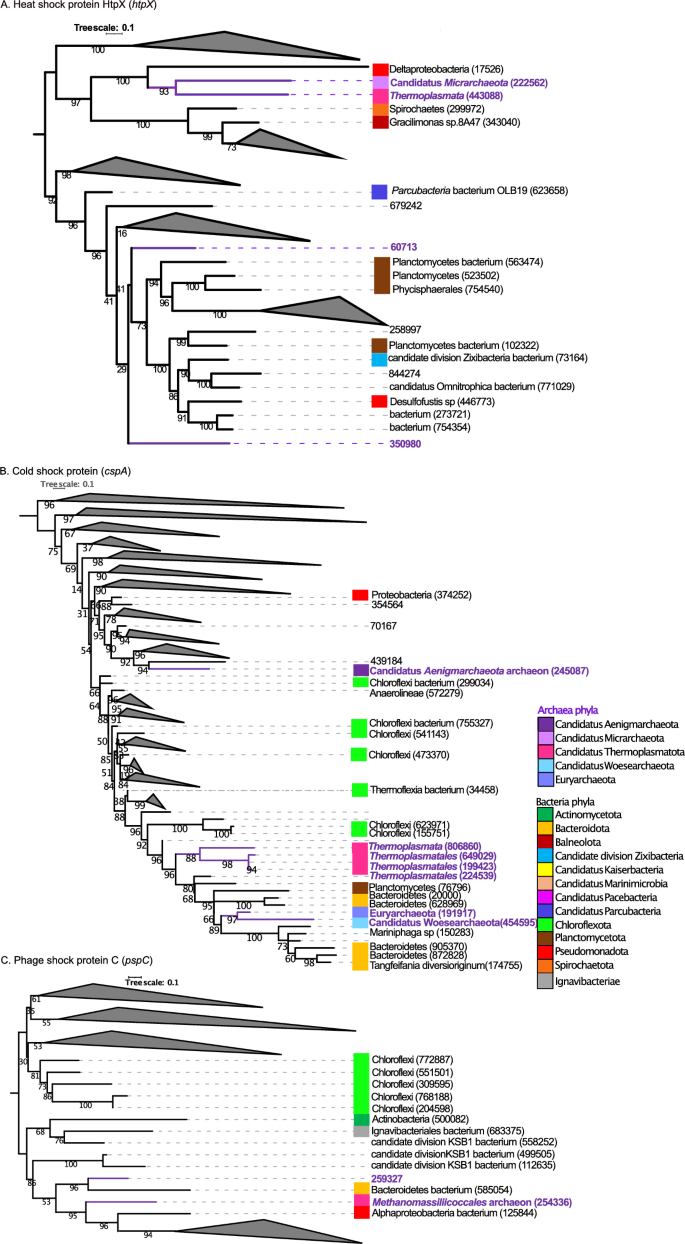
(A) Mid-point rooted phylogenetic tree primarily based on amino-acid sequences detected in our research from warmth shock protein HtpX (htpX). (B) Phylogenetic tree primarily based on amino-acid sequences from chilly shock protein (cspA). (C) Phylogenetic tree primarily based on amino-acid sequences from phage shock protein C (pspC). Amino-acid sequences from micro organism (black coloration), amino acid sequences from archaea (purple coloration). For constructed the tree, 454 sequences had been included within the phylogenetic evaluation (110 sequences for htpX gene, 228 sequences for cspA gene and 116 sequences for pspC gene). The sequences had been aligned with Muscle and the tree was generated with IQTREE2 with 1000 bootstraps, with auto-model choice through the built-in ModelFinder.
The phylogenetic bushes representing the UV genes (uvrA, uvrB, uvrC, uvrD, pcrA, uvsE, UVE1) are proven in Fig. 6 and within the supplementary Figs. S12–S15. For uvrA gene (Fig. 6A and S12), three genes categorized as coming from the archaeal lineages of Euryarchaeota and Thermoplasmata (genes IDs 848457, 361714, 636321) shaped a clade with a micro organism gene ID 519469 from Candidatus Buchananbacteria bacterium. One gene categorized as coming from Methanothermobacter (gene ID 727168) was inside a deep clade with bacterial genes from Phycisphaerae (gene ID 701638) and Brachyspira (gene ID 577760). For uvrB gene (Fig. S13), the gene ID 653100 taxonomically categorized as Thermoplasmata branched close to genes categorized as Planctomycetota (genes IDs 728796, 54766). For uvrC gene (Fig. 6B and S14), the genes IDs 699307, 905297, 647629 similar to archaea shaped a clade with 6 genes current in bacterial lineages: Pseudomonadota (gene ID 873573), Spirochaetes (gene ID 664616), Spirochaetia (gene ID 391558), Spirochaetia (gene ID 399873), Spirochaetia (gene ID 179402) and Gemmatimonadetes bacterium (gene ID 82391). For uvrD, pcrA gene (Fig. S15), the gene ID 181004 current in Candidatus Bathyarchaeota shaped a clade with a gene ID 297131 current in Bacteroidales bacterium. For uvsE, UVE1 gene (Fig. 6C), two genes current in Thermoplasmata (genes IDs 807563 and 510363) and one gene current in uncultured Caudoviricetes phage (gene ID 667218) shaped a clade with 3 genes current in micro organism (genes IDs 572443, 553365 and 74783, representing a Chloroflexi bacterium, an unclassified bacterium, and a Deltaproteobacteria bacterium, respectively).
[ad_2]
Source link


